Moraes MO, Silva LRB, Pinheiro RO. (2017) Chapter 6.1. Innate Immunity. In Scollard DM, & Gillis TP. (Eds.), International Textbook of Leprosy. American Leprosy Missions, Greenville, SC. https://doi.org/10.1489/itl.6.1
Introduction
Mycobacterium leprae is transmitted from an infected to a healthy person through aerosols containing bacteria. The most common route is likely to be the upper airways, indicating that interaction between M. leprae and the human host begins in the nasal mucosa. It is possible to detect M. leprae DNA in nasal swabs from healthy individuals in endemic areas [1], [2]. A follow-up of asymptomatic qPCR positive household contacts of leprosy patients using nasal swabs indicates that the air route is an important point of entry for M. leprae and, therefore, that these contacts have a higher chance of developing leprosy [3].
In the infection sites, resident macrophages and dendritic cells participate in the initial interaction between pathogen and host. Curiously, M. leprae has an exclusive ability to infect Schwann cells in peripheral nerves. All of these cells have a prominent role in the host immune response against infection with M. leprae, participating in processing and antigen presentation, cytokine secretion, and elimination of the pathogen.
Macrophages are capable of inducing inflammatory mediators that may activate specific lymphocyte subpopulations (see Chapter 6.2). This process establishes a profile of innate/adaptive immune responses that can restrict or permit the growth of infectious agents. Therefore, the initial interaction between M. leprae and macrophage processes is decisive for the outcome of the disease.
The immune response is dependent on genetic, environmental, and other factors that will ultimately induce macrophage activation that, in turn, may or may not commit the host cell towards effective antimicrobial activities. For example, a high bacilli count during infection can create a favorable niche for multiplication within macrophages [4]. In fact, in vitro assays have shown that M. leprae is able to maintain a stable metabolic state for a few weeks inside macrophages with a permissive phenotype when IL-10 is added to culture media and maintained at 33°C [5]. So, it is likely that a permissive program can be activated under certain conditions such as high bacillary numbers during infection and the presence of specific cytokines such as IL-10 [6]. Other environmental factors can also impact disease outcomes, such as nutrition and pregnancy [7], [8], [9]. The emergence of this type of permissive response is also dependent on genetic factors influencing the outcome of leprosy (see Chapter 8.1). It is worth reiterating that several genes encoding proteins involved in innate immunity have been associated with leprosy [10]. In this regard, well-known innate pathways can play a role in disease outcome, including the development of inflammatory reactions. These latter forms are immune-mediated altered clinical states (see Chapter 2.2).
In several aspects, our knowledge regarding potential mechanisms of M. leprae-induced innate immune activation comes from studies using other pathogenic mycobacteria. But, in recent years, advances in methodological approaches contributed to the understanding of immunopathogenic processes related to M. leprae infection. In the first part of this chapter, we describe the non-specific early mechanisms of host-pathogen interaction for M. leprae uptake by Schwann cells or macrophages. We also explore the regulation of cellular responses such as autophagy that culminate in a restrictive status that favors the killing of M. leprae. In addition, the subversion of these antimicrobial responses that induces a permissive environment for the bacteria is discussed. The second half of this chapter discusses the events of innate response and iron metabolism that regulate the secretion of cytokines, macrophage differentiation, activation of T cells, and adaptive immunity.
Early Events of Innate Immunity
M. leprae‒Host Cells interaction
Macrophages are present in almost all tissues, where they coordinate developmental, metabolic, and immunological functions and thus contribute to the maintenance of homeostasis. Macrophages provide a critical systemic network of cells to the innate immune system. They are major cytokine producers and are often placed to orchestrate not only innate but also adaptive immune responses [11]. Tissue macrophages are one of the main host cells for M. leprae, although in vitro experiments have shown that M. leprae is able to interact with keratinocytes promoting gene expression profile changes, increases in inflammatory cytokine, and cathelecidin production. Accordingly, the role of keratinocytes, in addition to macrophages, has been considered when evaluating the pathogenesis of leprosy [12], [13], [14]. Indeed, a comprehensive understanding of macrophage biology is crucial for grasping M. leprae pathogenesis.
M1 and M2 activated macrophages co-exist in adult tissue-based studies focused on primary macrophages and cell lines that are exposed in vitro to single, strongly polarized ligands such as lipopolysaccharide (LPS), interferon γ (IFN-γ), and interleukin 4 (IL-4). The term ‘classical activation’, which originally referred to macrophages stimulated with IFN-γ, is now used interchangeably to describe macrophages stimulated with IFN-γ and with TLR [15], [16]. In contrast, IL-4 is now described as inducing ‘alternative activation’, as compared to the effects of IFN-γ.
The initial phagocytosis of M. leprae is mediated by complement receptors CR1, CR3, and CR4 [17]. At the same time, M. leprae-macrophage interactions are mediated by pattern recognition receptors (PRRs) that recognize common molecular structures associated with pathogens (PAMPs). The interaction involves PRRs such as toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-containing protein 2 (NOD2). In this context, TLR2/1 heterodimer activation has been demonstrated to be crucial for M. leprae elimination. The mycobacterial synthetic lipoproteins (19 kDa and 33 kDa) increase pro-inflammatory cytokine responses in monocytes and dendritic cells mediated by TLR2/1 heterodimer [18]. This activation leads to an increase in both IL-12 and tumor necrosis factor (TNF) release. The latter of these is responsible for driving protective Th1-type responses, as an M1 macrophage phenotype is predominantly observed. It has been shown that the expression of TLR1 and TLR2 is up-regulated in skin biopsies of tuberculoid (TT) patients as compared to lepromatous (LL) patients [18]. Therefore, the activation of PRRs is key for stimulating antimicrobial responses. However, multiple TLRs participate during M. leprae infection that later trigger miRNA up-regulation as a feedback control. It is likely that TLR4 also participates in the M. leprae interaction, since miRNA-146a is one of the most highly induced miRNA by live M. leprae [19]. The up-regulation of miRNA-146a could be interpreted as a mechanism that the pathogen uses to circumvent the antimicrobial response [20]. In macrophage infection by other mycobacteria (BCG), miRNA-146a inhibits the activation of pro-inflammatory cytokines via down-regulation of IRAK1 and TRAF6 and induces the intracellular proliferation of bacteria [21].
Another important group of PRRs includes the nucleotide-binding oligomerization domain-like receptors called NLRs (or NOD-like receptors) that have been associated with the significant participation of the innate immune response during M. leprae infection [22]. In this context, NOD2 is a cytoplasmic sensor responsible for recognizing peptidoglycan, including mycobacterial derivatives, in which the main ligand is muramyl dipeptide (MDP) [23], [24]. NOD2 recognizes MDP and activates the transcription factor NF-kB through the adapter molecule RIP2, starting the production of pro-inflammatory mediators. Moreover, MDP can activate inflammation by caspase-1 recruitment, which is responsible for IL-1β cleavage and activation [25], [26]. NOD2 activation increases the autophagy pathway in dendritic cells and mediates antigen processing and presentation [27].
Regulation of autophagy and the Type I IFN pathway
Functional genomics and epidemiological studies have identified new pathways and candidate genes involved in leprosy susceptibility. One of these studies demonstrated that the E3 ubiquitin ligase, PARK2 (parkin), is a key player in trafficking mycobacteria to the autophagy pathway. Of potential relevance, PARK2 is a gene that has polymorphisms associated with leprosy in different populations [28] (see Chapter 8.1). Parkin has also been described as one of the proteins regulating autophagy. There is a great similarity between the process by which intracellular bacteria and mitochondria are marked for autophagy or mitophagy [29].
Autophagy is a critical cellular process for controlling cytosol cleansing and can also direct pathogens for phagolysosomal degradation. Indeed, parkin knockdown reduces autophagy and increases M. tuberculosis viability [30]. It is clear that autophagy plays a central role in the control of mycobacteria replication and spread. Several studies have dissected the pathways leading to activation of autophagy. M. tuberculosis induces selective autophagy mediated by ubiquitination in macrophage, resulting in the relocation of the bacilli to phagolysosomes. Other ubiquitin ligase proteins have been observed to cooperate with parkin in the formation of an autophagic complex in order to eliminate the mycobacteria [31]. Autophagy induced by M. tuberculosis is dependent on the ESX-1 secretion system and host cytosolic receptor STING (Stimulator of interferon genes) [30], [32], [33] that is triggered by type-I interferons.
Type I IFN-induced pathways are classically associated with antiviral responses, but they can also be up-regulated using DNA microarrays during live M. leprae infection [34], [35]. Although rare, leprosy and tuberculosis (TB) have been observed as side effects after treatment of hepatitis C with IFN-alpha [36], [37]. Moreover, other global gene expression studies point to the importance of this pathway in TB progression [38]. Indeed, an important family member of interferon-stimulated genes is 2’–5’ oligoadenylate synthetase like (OASL), which has also been described as induced during progression from latent to active TB [39]. OASL was strongly induced in Schwann cells and human macrophages during M. leprae infection, suggesting that this gene is an early sign of infection with virulent mycobacteria [35]. The pathway that induces type I IFN is also dependent on the ESX-1/ESAT-6 mycobacterial secretion system and the cGAS/STING pathway [32]. In addition, type I IFN induces OASL expression [35] and, when it is knocked down, a large decrease in the viability of mycobacteria and increased autophagy, thereby suggesting that the OASL participates in the regulation of an immune response that diverts antimicrobial responses. One of the readouts of this kind of response is CCL2/MCP-1, which is secreted in high amounts when a permissive phenotype is observed and is down-regulated when OASL is silenced [35]. Curiously, parkin knockdown also decreased CCL2/MCP-1 and IL-6 production in cells stimulated with live M. leprae [40].
Thus, mechanisms that trigger Type I IFN and parkin are related to pathways induced upon M. leprae infection. But it remains unclear as to how the interaction of macrophages with M. leprae leads to autophagy, or to a permissive state that allows M. leprae a safe intracellular niche. Another study has provided evidence that the permissive phenotype, largely observed in disseminated LL skin lesions, is indeed due to reduction in the activation of autophagy, decreasing macrophage antimicrobial activity [41]. These data suggest that the inhibition of autophagy activation mediated by mycobacteria, and possibly involving down-regulation of parkin, constitutes a crucial immunological mechanism associated with control of disease and polarization towards disseminated forms of the disease.
In fact, a number of other simultaneous signaling processes involving pattern recognition receptors, such as NOD2 and TLR1/2, also participate in the response of inflammatory macrophages against infection by virulent mycobacteria. Genetic variations associated with susceptibility to leprosy suggest subtle changes in expression levels of genes in the type I IFN pathway, lipid metabolism, energy metabolism, and autophagy that could impact the disease outcome (see Chapter 8.1). In this context, lipid bodies (LBs), which are considered lipid-rich organelles, may also play an important role in regulating autophagy [42]. The location of ubiquitination system components inside these lipid bodies (LB) suggests that these processes are actually interconnected in the regulation of innate immune responses to M. leprae.
Metabolism modulation
Cholesterol is among the lipids with increased abundance in M. leprae-infected cells, but its essential role for M. leprae intracellular survival does not rely on its utilization as a nutritional source [43], [44]. Even though they do not use cholesterol as a nutrient, macrophages infected with M. leprae have a greater capacity to synthesize and sequester exogenous cholesterol, contributing to bacterial intracellular survival. The analysis of LL macrophages has revealed an increased expression of host lipid metabolism genes [45]. In this context, lipid body formation induced by M. leprae in the host cell has been associated with the pathogenesis of leprosy contributing to persistent infection [46]. M. leprae decreases hormone sensitive lipase (HSL), facilitating the maintenance of a lipid-rich environment suitable for intracellular survival of M. leprae. Studies in vitro have demonstrated that live M. leprae has the ability to actively induce and support adipophilin/adipose differentiation related protein/peripilin (ADRP) expression to facilitate the accumulation of lipids within the phagosome [47]. LBs are transported to lysosomes via autophagic complex formation, where LB hydrolysis occurs to generate free cholesterol efflux mainly dependent on ABCA1 in foamy macrophages. This process mobilizes cholesterol associated with LB for reverse cholesterol transport [48]. Clofazimine, used in multibacillary (MB) multi-drug therapy (MDT), attenuates ADRP in M. leprae-infected cells and increases HSL [49], [50]. Finally, statins, which are drugs that inhibit cholesterol synthesis, have demonstrated efficacy as an adjuvant to controlling the infection. The statins enhanced the antimicrobial effect of rifampin in M. leprae-infected mice, demonstrating that cholesterol metabolism modulation may play an important role in the pathogenesis of leprosy [51].
M. leprae has experienced reductive evolution at the genome level (see Chapter 8.2), which resulted in the loss of essential genes of bacterial metabolism associated with energy metabolism. Thus the bacteria became highly dependent upon host cell resources for its survival. This strategy is demonstrated when M. leprae enhances G6PDH enzyme activity, and increases glucose uptake in Schwann cells, redirecting cell metabolism towards the pentose phosphate pathway [52]. This strategy switches cellular responses to lipid synthesis and glutathione antioxidant system maintenance, suggesting a Warburg-like phenomenon. Also, a decrease in lactate release in a culture medium of M. leprae-infected Schwann cells can lead to lactate restriction for the axon. This lactate restriction may be involved in neurodegenerative and demyelination processes, both of which are observed in leprosy [50].
Metabolic pathway regulation has been shown to play a crucial role in leprosy pathogenesis. Metabolomic analysis shows that LL patients have altered metabolism when compared to healthy subjects [51]. In fact, the patient’s metabolic profile is restored after treatment. The altered profile mainly consists of polyunsaturated fatty acids with pro-inflammatory resolvins and higher levels of lipids derived from lipase activity. Modulation of polyunsaturated lipids as omega-3 and -6 has been observed during the course of M. leprae infection. Also, high levels of PGD2, PGE2, lipoxin A4, and resolvins in skin lesions of LL patients have been reported [53].
Indeed, energetic metabolism can be orchestrated by growth factors. M. leprae infection is able to increase the expression and secretion of insulin-like growth factor-I (IGF-I), a classic hormone that exhibits anti-apoptotic and proliferative activities in Schwann cells and macrophages. Macrophages from dermal lesions of LL patients may show a higher expression of IGF-I than those from the self-limited borderline tuberculoid (BT) leprosy form. In vitro studies have demonstrated that the dampening of IGF-I signaling reverts the capacity of M. leprae-infected human and murine macrophages to produce antimicrobial molecules and promotes bacterial killing [54], [55], [56]. The inhibition of antimicrobial activity in LL patients is in accordance with in vivo observations in which a higher SOCS3 expression and lower phosphorylation of STAT1 levels were found in LL versus BT dermal lesions [54], [55]. An overview of the most important mediators and processes involved in the early events of the immune response after M. leprae infection is shown in Figure 1.

In a first step of host-bacteria interaction, M. leprae is phagocytosed by macrophages using complement receptors and then the mycobacteria cell wall is recognized by TLR2, inducing lipid body formation [17], [57]. The phagocytosed M. leprae can breach the phagosome membrane in a bacterial ESX-1 secretion system-dependent manner.
This event leads to bacterial DNA release into the cytosol, activating the cGAS/STING/IRF3 pathway and increasing type I IFN secretion-inhibiting autophagy by a mechanism dependent on OASL activation [35], [58]. Also, M. leprae-infected macrophages increase glucose uptake, IGF-I expression, and G6PD expression [52] that, along with OASL, favor mycobacterial survival [35], [54]. All of these events characterize a permissive program that culminates with a successful infection [52].
In contrast, antimicrobial routes can be induced in order to clear bacteria. In this case, the cell activates the ubiquitin ligases Parkin and Smurf1 that are responsible for ubiquitination of the bacilli (or an intermediary host protein that ligates the bacteria), targeting the bacteria for degradation in the autophagosome. Also, vitamin D activates the VDR transcription factor that is responsible for increased levels of the antimicrobial peptide cathelicidin (CAMP) [6].
| Schwann cell invasion by M. leprae |
| The interaction of M. leprae with Schwann cells (SC) is mediated by adhesins present in the cell wall of the bacillus that interact with the host cell extracellular matrix. The M. leprae HLP (histone-like protein) protein, PGL-I, and other cell wall components have been described as ligands of the M. leprae wall that are capable of interacting with the G domain of the α-2 laminin chain extracellular matrix protein of the host. Indeed, the α-2 laminin chain functions as a cellular receptor for the bacillus, promoting its interaction with the α-dystroglican present in the basal lamina that surrounds the SC, leading to internalization of the pathogen [59], [60], [61]. Furthermore, GD3 ganglioside is also involved in adhesion and internalization during M. leprae infection [62]. |
| The interaction of M. leprae with Schwann cells (SC) is mediated by adhesins present in the cell wall of the bacillus that interact with the host cell extracellular matrix. The M. leprae HLP (histone-like protein) protein, PGL-I, and other cell wall components have been described as ligands of the M. leprae wall that are capable of interacting with the G domain of the α-2 laminin chain extracellular matrix protein of the host. Indeed, the α-2 laminin chain functions as a cellular receptor for the bacillus, promoting its interaction with the α-dystroglican present in the basal lamina that surrounds the SC, leading to internalization of the pathogen [59], [60], [61]. Furthermore, GD3 ganglioside is also involved in adhesion and internalization during M. leprae infection [62]. |
| M. leprae infection promotes SC demyelination mediated by direct bacterial binding and ErbB2 receptor activation [63]. It has been demonstrated that during M. leprae infection, human primary SC undergo a cellular reprogramming that leads to an immature progenitor/stem cell-like cell (pSLC) phenotype. Before de-differentiation, SCs are highly infected, and right after the reprogramming step, pSLCs lose bacilli retention capacity, acquiring a high bacterial transfer capacity to other cells, such as fibroblasts. pSLCs facilitate the spread of infection to other tissues, creating a bacilli flow that enables a sustained infection [64]. |
Late Events of Innate Immunity
Iron metabolism: Regulation of innate responses and M. leprae survival
Mycobacteria require iron for growth and virulence. Iron is an essential nutrient for the survival of organisms, and pathogenic bacteria have developed sophisticated pathways to satisfy their iron requirements. With some exceptions like Borrelia burgdorferi, which uses manganese in place of iron, most pathogenic bacteria require iron to maintain essential pathways like electron transport, antioxidant defense, synthesis of aromatic amino acids, and other functions [65]. Free iron is toxic to host cells. Therefore, both host and bacteria tightly regulate iron and heme release. Regulation of iron metabolism involves sensing intracellular iron concentrations and modulating uptake and storage accordingly. Iron storage proteins are important to protect against oxidative stress by sequestering ferrous iron and consequently limiting toxic hydroxyl radical (OH‒) formation by Fenton chemistry [66], [67].
Since little unchelated iron exists within the host, invading bacteria must obtain their iron from circulating iron-containing proteins, such as transferrin or lactoferrin, or from the iron-storage protein ferritin. Mycobacteria synthesize low molecular mass molecules called siderophores. Due to higher affinity, siderophores may remove iron from these molecules [68] and provide a means for iron acquisition, leading to mycobacteria growing in vitro and in vivo. Mycobacteria produce two classes of siderophores: mycobactins and exochelins [69], [70], [71], [72]. Mycobactins are found in two forms that differ in polarity and solubility. The less polar form remains cell-associated (mycobactin), and the more polar one (carboxymycobactin) is secreted into the medium. Intraphagosomal mycobacteria must induce the synthesis of siderophores in order to acquire iron from transferrin but, when sufficient iron has been acquired, regulatory pathways may down-regulate iron uptake in response to the iron-sufficient conditions that they may now face [73].
Microbial iron uptake seems to occur as follows. Soluble siderophores are secreted to obtain iron from the environment or from the host and are transformed into ferric siderophores. These ferric siderophores are then transported into cells by receptors located in the cell envelope. Next, iron ions are delivered to bacterioferritin to satisfy the physiological requirements. Both exochelins and carboxymycobactins can seize the extracellular iron and transfer iron to the intracellular siderophores [74].
M. leprae does not contain mycobactin (see Chapter 8.2). This lack suggests that M. leprae might be considered a mycobactin-dependent microbe and that other mycobacteria present in humans might provide sufficient mycobactin for in vivo multiplication of M. leprae. M. leprae does produce several proteins involved in iron transport, storage, and regulation in the absence of recognizable genes encoding iron scavengers [75].
The complete annotation of the M. leprae genome includes two bacterioferritin genes [76]. One of these genes (ML2038) encodes the 18 kDa BfrA, which corresponds to the previously identified M. leprae bacterioferritin [77]. The second is a pseudogene (ML0075) which encodes the ferritin-like protein BfrB. MDP-1 from M. leprae (Mycobacterial DNA-binding protein 1: ML1683; ML-LBP) has been described as a histone-like protein that is a ferroxidase-iron storage protein outside of the ferritin superfamily proteins [78].
Iron requirements in M. leprae-infected host cells
The major pathway for eukaryotic cells to avoid toxicity from intracellular iron accumulation is the translocation of Fe+2 into the cytosol. There, it is safely stored in the oxidized Fe+3 form bound to ferritin, a function that is in large part performed by the divalent cation efflux pump Nramp2 (slc11a2/DMT1). Macrophages express an additional metal transporter, Nramp1 (slc11a1), in their late endosomes/lysosomes. Nramp1 is expressed in the lysosomal compartment, whereas Nramp2 is not detectable in the lysosomes but is expressed primarily in recycling endosomes and also, to a lesser extent, at the plasma membrane, colocalizing with transferrin. Nramp2 transports free Fe2+ across the endosomal membrane and into the cytoplasm [79], [80]. Authors have hypothesized that the phagocyte-specific Nramp1 protein may regulate the intraphagosomal replication of mycobacteria by controlling divalent cation concentrations at that site [81].
Iron-mediated anti-mycobacterial defenses of the host may follow two pathways. The first is to sequester mycobacterial molecules that have already captured iron. For example, siderocalin (Lcn 2), has been shown to bind to soluble siderophores of mycobacteria [82], thus preventing the utilization of the iron by the pathogen.
The second pathway is to make iron unavailable to mycobacteria. Hepcidin is an antimicrobial peptide produced by hepatocytes in response to excess iron or inflammation and is one of the principal iron regulatory hormones [83]. Hepcidin orchestrates an innate immune response to further reduce available iron and to slow or stop the growth of bacteria [84], [85], [86]. Hepcidin stimulates iron accumulation inside cells by inducing the internalization and destruction of ferroportin, a plasma membrane-associated iron transporter protein (see Table 1) that regulates the export of iron from duodenal enterocytes and macrophages into plasma in the human host [65], [87], [88]. Hepcidin has been considered a link between inflammation and the anemia of chronic diseases. The latter is characterized by reduced levels of iron in the serum and increased iron reserves, as evidenced by elevated ferritin and several inflammatory mediators that may regulate hepcidin expression in host cells, like IL-6 and IL-1β. Bacterial infection of macrophages may also drive hepcidin production via TLR activation [89].
|
Protein |
Role in Iron Metabolism |
Role in Immune Response or Inflammation |
Influence on M. leprae Infection |
|
Binds directly to the iron efflux protein ferroportin and contributes to the depletion of circulating iron that would be available to extracellular pathogens. |
Upon activation of PPRs, secreted IL-6 induces hepcidin expression and release from the liver. Hepcidin activates the protein kinase Jak2, which phosphorylates the transcription factor Stat3, which may contribute to the modulation of downstream acute cytokine-induced inflammatory responses. |
Disruption of the hepcidin/ferroportin axis has an impact on leprosy immunopathology. Paucibacillary (PB) macrophages have higher expression of ferroportin, whereas MB cells have increased hepcidin and decreased ferroportin expression. |
|
|
A potent chelator of ferric iron that is involved in the modulation of extracellular iron availability. |
The exact role of lactoferrin in innate immunity is not fully elucidated, but it is known that TNF may induce the release of lactoferrin from neutrophilic granules at the site of a bacterial infection. |
Serum lactoferrin is strongly and inversely associated with increasing BI. It needs to be evaluated, but since increased systemic TNF is observed in patients with leprosy, it can be hypothesized that lactoferrin is released by neutrophils in infection sites to sequester iron. |
|
|
Subverts the iron acquisition system through binding to salicylate-derived iron-laden siderophores such as mycobactin secreted by M. tuberculosis. |
Produced by neutrophil granules and in epithelial cells in response to IL-1β. |
It probably limits iron availability to M. leprae, as described for M. tuberculosis. |
|
|
Transferrin receptors [96] |
Its down-regulation limits the cell’s ability to acquire transferrin-bound iron and reduces the endosomal pool of iron accessed by intracellular pathogens. |
Production of nitric oxide by LPS/IFN-γ-activated macrophages may down-regulate transferrin receptors and up-regulate the synthesis of ferritin. |
Down-regulation during infection reduces endosomal iron to restrict pathogen growth. |
|
Ferroportin |
Membrane exporter of iron. It opposes the systemic regulation by hepcidin described above. |
Cytokines that regulate hepcidin expression may affect the hepicidin/ferroportin axis, e.g., IL-6 and IL-1β. |
Decreased expression in lepromatous macrophages contributes to the retention of iron inside cells and concomitantly aids in pathogen survival. |
|
Nramp1 |
Reduces intraphagosomal iron. |
The mobilization of iron by Nramp1 suppresses the expression of IL-10 and enhances macrophage production of iNOS. |
It may confer resistance to M. leprae. |
The evaluation of hepcidin in patients with the polar forms of leprosy has demonstrated that hepcidin is elevated in LL lesions, whereas ferroportin is more abundant in TT ones [97]. Anemia is a common finding in patients presenting with the MB form of leprosy [98], [99], [100], [101]. Indeed, in MB patients, changes in hematologic parameters indicating altered iron metabolism appeared to result from a mixture of anemia from inflammation and iron deficiency [91]. Independent of the clinical parameters, the analysis of macrophages present in skin lesions from leprosy patients demonstrated increased iron storage in host cells from LL but not from TT patients, suggesting that iron sequestration inside host cells might provide an optimal environment for the bacillus [102].
The increased iron storage in LL cells (Figure 2) may be due not only to the decreased expression of ferroportin, but also to an increase in CD163 expression. Previous studies demonstrated that the degradation of senescent erythrocytes by macrophages produces heme that may be utilized as a source of iron by M. tuberculosis [103], [104]. In leprosy, other mechanisms related to iron acquisition for the bacilli have been described. CD163, a receptor of hemoglobin and hemoglobin-haptoglobin (Hb-Hp) complexes, is highly expressed in cells from LL lesions when compared with TT lesions. Because CD163 is related to hemoglobin clearance, it can be speculated that in infected cells, high CD163 expression may function as a pathway for the supply of iron. The increase in CD163 expression in LL macrophages is accompanied by an increase in iron storage [102], which reinforces the hypothesis that iron is important for M. leprae survival inside host cells and suggests that heme may be a source of iron for M. leprae as well.
 lesions have increased iron storage in the form of ferritin and hemosiderin as compared to borderline tuberculoid patients (BT). Hemosiderin deposition was analyzed by the Prussian Blue method. Expression of the protein ferritin light chain was assessed by immunohistochemical analysis.")
A primary question remains as to how iron is internalized by the mycobacteria inside the phagosome. One possibility is that lipid bodies may contribute to the delivery of iron to the phagosome. As described earlier, M. leprae induces several changes in the host cell so that it becomes an advantageous niche for growth. Indeed, the production of lipid bodies could also help with iron uptake [46], [105]. Others have demonstrated that, after bacterial infection, LBs within human macrophages concentrate into phagosomes and change the phagosomal contents [106], [107]. It is hypothesized that mycobacteria may benefit from this host defense mechanism to deliver iron during infection [108]. Mycobactins may diffuse out of mycobacterial-loaded phagosomes within macrophages and become associated with either the intracellular iron pool or migrate to the macrophage plasma membrane for iron chelation. These iron-loaded mycobactins may accumulate within lipid storage and sorting vesicles of the host macrophage. The lipid bodies, carrying iron-loaded mycobactins, may remain in direct contact with mycobacteria-containing phagosomes and are thought to be involved in the delivery of the loaded iron inside the phagosomes, whereby the enriched phagosomal iron pool may be used by the bacteria [108].
Macrophage program modulation by M. leprae infection
The interaction of M. leprae with the macrophage and M. leprae’s ability to trigger a genetic, metabolic, and differentiation program will define the cellular commitment towards a permissive or a restrictive phenotype. The very early interaction triggered upon infection described in the beginning of this chapter (Type I IFNs, autophagy, iron uptake) culminates in effector responses such as cytokine secretion and macrophage phenotype differentiation. Generally, the profile of the specific immune response is a factor in distinguishing between the different clinical forms of leprosy. A combination of genetic and environmental factors contribute to the overall cellular phenotype that leads to a more disseminated form of the disease, LL, or to a more localized form, TT.
For example, IL-10 induces a phagocytic differentiation program resulting in the up-regulation of scavenger receptor CD163, which may function as a co-receptor for M. leprae entry in host cells [102]. In addition, IL-10 increases the phagocytic capacity of macrophages. In LL lesions, the predominant subset of macrophages exhibits high expression of CD163. CD163 and foamy phenotype in LL macrophages are associated with increased iron deposition that contributes to M. leprae survival inside host cells [102].
On the other hand, IL-15 leads to the activation of a vitamin D-dependent antimicrobial program, along with a reduced phagocytic capacity. Macrophages with an antimicrobial profile are predominantly found in the lesions of TT patients [6]. The vitamin D mechanism involves induction of the CYP27B1 enzyme, which is responsible for converting the 25-hydroxyvitamin D to its biologically active form, 1,25 hydroxyvitamin D; positive regulation of the vitamin D receptor (VDR); and increased expression of cathelicidin, an important antimicrobial peptide [109], [110], [111]. At the same time, VDR activation, along with the production of IL-1β, is capable of inducing defensin B (DEFB4), another peptide required for antimicrobial activity of the host cell [112].
In fact, tissue macrophage analysis from leprosy patients has revealed that in polar LL disease, there is an increase of M2 markers, including CD68, CD163, arginase 1, IL-10, IL-13, TGF-β, and FGF-b [113]. Galectin-3 contributes to IL-10 expression and is increased in LL macrophages [114]. In vitro studies have demonstrated that live M. leprae may induce an M2-like macrophage function by decreasing IL-1β, IL-6, TNF, and MHC II expression and increasing both CD163 and IL-10 expression [115]. When incubated with naïve T cells, macrophages treated with live M. leprae preferentially primed regulatory T (Treg) cell responses with elevated FoxP3 and IL-10 expression, while reducing interferon gamma (IFN-gamma) expression and CD8+ T cell cytotoxicity [115].
Although a predominance of permissive macrophages has been observed in LL lesions, the use of terms such as “suppressive” or “M2 cells” might be an oversimplification, since several lines of evidence suggest the presence of both pro- and anti-inflammatory molecules in LL macrophages. For example, solute carrier family 11a member 1 (Slc11a1) and inducible Nitric Oxide Synthase (iNOS) play an important role in the control of intracellular pathogens, but are positively correlated with the bacillary load in LL macrophages [116], demonstrating that the LL phenotype occurs independently of these “protective” molecules. IDO1 is an enzyme that may have a dual role, acting both in regulatory and pro-inflammatory pathways. Previous studies have demonstrated that IDO1 expression is increased in LL macrophages [117] and, on the other hand, may be increased in an antimicrobial context to control the high bacillary load or, finally, in a negative feedback regulatory axis to increase anti-inflammatory cytokines and contribute to disease persistence [102].
Despite the fact that LL macrophages present classical M2 markers and anti-inflammatory cytokines, iron retention, a marker of M1 cells, is a common finding in LL cells [102]. This finding suggests that LL cells may have a phenotype that may not be classified in the paradigmatic M1-M2 profile (Figure 3).
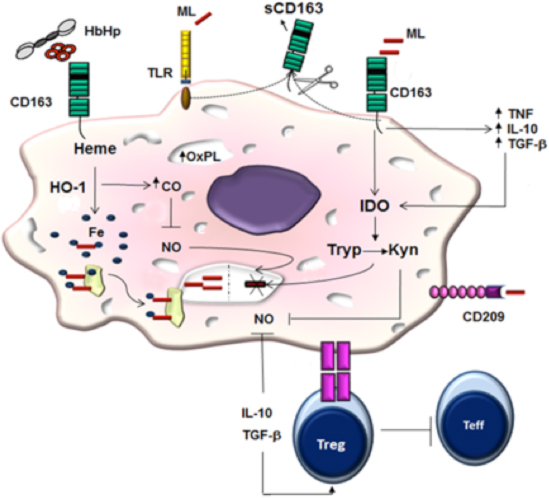
M. leprae is a poor activator of macrophages and dendritic cells in vitro. In naïve human monocytes, M. leprae induces CCL-2 and IL-1ra while suppressing IL-6 production through PI3K-dependent mechanisms [121]. Although M. leprae is a weak stimulator of cytokines, it has been shown to prime human monocytes for the increased production of TNF and IL-10 in response to a strongly inducing secondary stimulus [121]. IL-10 expression is also correlated with IL-27, a cytokine that inhibits the capacity of IFN-γ to trigger antimicrobial activity against M. leprae in infected monocytes [122].
Macrophages have evolved a myriad of defense strategies to cope with intracellular bacteria [123], and the understanding of immune mechanisms associated with the paucibacillary (PB) form of the disease may reveal new perspectives in deciphering the crucial pathways related to protection in leprosy. In this sense, it has been demonstrated that the phagocytosis of apoptotic cells (efferocytosis) increases M. leprae survival in differentiated M1 cells (human CD14+ cells differentiated with GM-CSF), accompanied by reduced IL-15 and IL-6 levels and increased TGF-β and IL-10 secretion. GM-CSF-differentiated cells primed with M. leprae in the presence of apoptotic cells induced the secretion of Th2 cytokines IL-4 and IL-13 in autologous T cells compared with cultures stimulated with M. leprae or apoptotic cells alone. Together, these data suggest that in PB leprosy patients, efferocytosis contributes to mycobacterial persistence by inducing a population of macrophages more susceptible to infection. In this case, GM-CSF-differentiated cells in the presence of M. leprae and apoptotic cells displayed an increased CD163 and SRA-I expression as well as a higher phagocytic capacity when compared with non-stimulated cells [124].
Conclusions
Mycobacterium leprae is able to subvert innate immunity, triggering cellular differentiation of macrophages towards a permissive phenotype for mycobacterial growth and survival. This mechanism requires activation of Type I IFNs and a shift in energy metabolism shutting down mitochondria and increasing the pentose phosphate pathway directing lipid synthesis. This phenotype decreases parkin, autophagic mechanisms, and other antimicrobial pathways, resulting in disease progression. The establishment of this permissive phenotype depends on iron sequestration and CD163 as a scavenging receptor, which creates a microenvironment with secretion of IL-10 and CCL2 and decreased production of IL-15 and IL-1-beta. To sum up, in PB clinical forms, macrophages display a microbicidal phenotype, with antimicrobial responses mediated by the vitamin D pathway that contributes to an increase in antimicrobial peptides and also the up-regulation of autophagy. In MB forms, macrophages display a permissive phenotype that contributes to bacterial persistence.
Indeed, early events in M. leprae interaction with macrophages and Schwann cells involve several biochemical and immunological pathways. An The increased definition of these pathways may provide novel pharmacological targets that could be use as adjuncts to MDT, thereby improving care for leprosy patients.
Footnotes
- ^ Klatser PR, van Beers S, Madjid B, Day R, de Wit MY. 1993. Detection of Mycobacterium leprae nasal carriers in populations for which leprosy is endemic. J Clin Microbiol 31:2947–2951.
- ^ Patrocínio LG, Goulart IM, Goulart LR, Patrocínio JA, Ferreira FR, Fleury RN. 2005. Detection of Mycobacterium leprae in nasal mucosa biopsies by the polymerase chain reaction. FEMS Immunol Med Microbiol 44:311–316.
- ^ Araujo S, Freitas LO, Goulart LR, Goulart IMB. 2016. Molecular evidence for the aerial route of infection of Mycobacterium leprae and the role of asymptomatic carriers in the persistence of leprosy. Clin Infect Dis 63:1412–1420.
- ^ Hagge DA, Ray NA, Krahenbuhl JL, Adams LB. 2004. An in vitro model for the lepromatous leprosy granuloma: fate of Mycobacterium leprae from target macrophages after interaction with normal and activated effector macrophages. J Immunol 172:7771–7779.
- ^ Fukutomi Y. 2004. [Functional changes of macrophages in Hansen’s disease]. Nihon Hansenbyo Gakkai Zasshi 73:253–261.
- a, b, c Montoya D, Cruz D, Teles RM, Lee DJ, Ochoa MT, Krutzik SR, Chun R, Schenk M, Zhang X, Ferguson BG, Burdick AE, Sarno EN, Rea TH, Hewison M, Adams JS, Cheng G, Modlin RL. 2009. Divergence of macrophage phagocytic and antimicrobial programs in leprosy. Cell Host Microbe 6:343–353.
- ^ Saha K, Rao KN, Chattopadhya D, Lakshmi V, Gadi S, Dutta Banik ND. 1990. A study on nutrition, growth and development of a high-risk group of children of urban leprosy patients. Eur J Clin Nutr 44:471–479.
- ^ Feenstra SG, Nahar Q, Pahan D, Oskam L, Richardus JH. 2011. Recent food shortage is associated with leprosy disease in Bangladesh: a case-control study. PLoS Negl Trop Dis 5:e1029.
- ^ Ozturk Z, Tatliparmak A. 2017. Leprosy treatment during pregnancy and breastfeeding: a case report and brief review of literature. Dermatol Ther 30.
- ^ Cardoso CC, Pereira AC, de Sales Marques C, Moraes MO. 2011. Leprosy susceptibility: genetic variations regulate innate and adaptive immunity, and disease outcome. Future Microbiol 6:533–549.
- ^ Wynn TA, Chawla A, Pollard JW. 2013. Macrophage biology in development, homeostasis and disease. Nature 496:445–455.
- ^ Cogen AL, Walker SL, Roberts CH, Hagge DA, Neupane KD, Khadge S, Lockwood DN. 2012. Human beta-defensin 3 is up-regulated in cutaneous leprosy type 1 reactions. PLoS Negl Trop Dis 6:e1869.
- ^ Polycarpou A, Walker SL, Lockwood DN. 2013. New findings in the pathogenesis of leprosy and implications for the management of leprosy. Curr Opin Infect Dis 26:413–419.
- ^ Lyrio EC, Campos-Souza IC, Corrêa LC, Lechuga GC, Verícimo M, Castro HC, Bourguignon SC, Côrte-Real S, Ratcliffe N, Declercq W, Santos DO. 2015. Interaction of Mycobacterium leprae with the HaCaT human keratinocyte cell line: new frontiers in the cellular immunology of leprosy. Exp Dermatol 24:536–542.
- ^ Stein M, Keshav S, Harris N, Gordon S. 1992. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med 176:287–292.
- ^ Martinez FO, Gordon S. 2014. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000prime Rep 6:13.
- a, b Schlesinger LS, Horwitz MA. 1991. Phagocytosis of Mycobacterium leprae by human monocyte-derived macrophages is mediated by complement receptors CR1 (CD35), CR3 (CD11b/CD18), and CR4 (CD11c/CD18) and IFN-gamma activation inhibits complement receptor function and phagocytosis of this bacterium. J Immunol 147:1983–1994.
- a, b Krutzik SR, Ochoa MT, Sieling PA, Uematsu S, Ng YW, Legaspi A, Liu PT, Cole ST, Godowski PJ, Maeda Y, Sarno EN, Norgard MV, Brennan PJ, Akira S, Rea TH, Modlin RL. 2003. Activation and regulation of Toll-like receptors 2 and 1 in human leprosy. Nat Med 9:525–532.
- ^ Cezar-de-Mello PF, Toledo-Pinto TG, Marques CS, Arnez LE, Cardoso CC, Guerreiro LT, Antunes SL, Jardim MM, Covas Cde J, Illaramendi X, Dias-Baptista IM, Rosa PS, Durães SM, Pacheco AG, Ribeiro-Alves M, Sarno EN, Moraes MO. 2014. Pre-miR-146a (rs2910164 G>C) single nucleotide polymorphism is genetically and functionally associated with leprosy. PLoS Negl Trop Dis 8:e3099.
- ^ Liu PT, Wheelwright M, Teles R, Komisopoulou E, Edfeldt K, Ferguson B, Mehta MD, Vazirnia A, Rea TH, Sarno EN, Graeber TG, Modlin RL. 2012. MicroRNA-21 targets the vitamin D-dependent antimicrobial pathway in leprosy. Nat Med 18:267–273.
- ^ Li S, Yue Y, Xu W, Xiong S. 2013. MicroRNA-146a represses mycobacteria-induced inflammatory response and facilitates bacterial replication via targeting IRAK-1 and TRAF-6. PLoS One 8:e81438.
- ^ Schenk M, Mahapatra S, Le P, Kim HJ, Choi AW, Brennan PJ, Belisle JT, Modlin RL. 2016. Human NOD2 recognizes structurally unique muramyl dipeptides from Mycobacterium leprae. Infect Immun 84:2429–2438.
- ^ Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. 2003. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 278:8869–8872.
- ^ Kim TH, Chae GT, Kang TJ. 2015. Expression of Nucleotide-Oligomerization Domain (NOD) and related genes in mouse tissues infected with Mycobacterium leprae. Immune Netw 15:319–324.
- ^ Delbridge LM, O’Riordan MX. 2007. Innate recognition of intracellular bacteria. Curr Opin Immunol 19:10–16.
- ^ Kang TJ, Chae GT. 2011. The role of intracellular receptor NODs for cytokine production by macrophages infected with Mycobacterium leprae. Immune Netw 11:424–427.
- ^ Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A. 2010. NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nat Med 16:90–97.
- ^ Mira MT, Alcaïs A, Nguyen VT, Moraes MO, Di Flumeri C, Vu HT, Mai CP, Nguyen TH, Nguyen NB, Pham XK, Sarno EN, Alter A, Montpetit A, Moraes ME, Moraes JR, Doré C, Gallant CJ, Lepage P, Verner A, Van De Vosse E, Hudson TJ, Abel L, Schurr E. 2004. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 427:636–640.
- ^ Deretic V. 2010. Autophagy in infection. Curr Opin Cell Biol 22:252–262.
- a, b Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS. 2013. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501:512–516.
- ^ Franco LH, Nair VR, Scharn CR, Xavier RJ, Torrealba JR, Shiloh MU, Levine B. 2017. The ubiquitin ligase Smurf1 functions in selective autophagy of Mycobacterium tuberculosis and anti-tuberculous host defense. Cell Host Microbe 21:59–72.
- a, b Manzanillo PS, Shiloh MU, Portnoy DA, Cox JS. 2012. Mycobacterium tuberculosis activates the DNA-dependent cytosolic surveillance pathway within macrophages. Cell Host Microbe 11:469–480.
- ^ Watson RO, Manzanillo PS, Cox JS. 2012. Extracellular M. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150:803–815.
- ^ Guerreiro LT, Robottom-Ferreira AB, Ribeiro-Alves M, Toledo-Pinto TG, Rosa Brito T, Rosa PS, Sandoval FG, Jardim MR, Antunes SG, Shannon EJ, Sarno EN, Pessolani MC, Williams DL, Moraes MO. 2013. Gene expression profiling specifies chemokine, mitochondrial and lipid metabolism signatures in leprosy. PLoS One 8:e64748.
- a, b, c, d, e, f de Toledo-Pinto TG, Ferreira AB, Ribeiro-Alves M, Rodrigues LS, Batista-Silva LR, Silva BJ, Lemes RM, Martinez AN, Sandoval FG, Alvarado-Arnez LE, Rosa PS, Shannon EJ, Pessolani MC, Pinheiro RO, Antunes SL, Sarno EN, Lara FA, Williams DL, Ozório Moraes M. 2016. STING-dependent 2’–5’ oligoadenylate synthetase-like production is required for intracellular Mycobacterium leprae survival. J Infect Dis 214:311–320. doi:10.1093/infdis/jiw144
- ^ Velasco J, González S. 2011. [Tuberculous lymphadenitis after successful treatment of hepatitis C]. Enferm Infecc Microbiol Clín 29:631.
- ^ Santos M, Franco Edos S, Ferreira PL, Braga WS. 2013. Borderline tuberculoid leprosy and type 1 leprosy reaction in a hepatitis C patient during treatment with interferon and ribavirin. An Bras Dermatol 88:109–112.
- ^ Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O’Garra A. 2010. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466:973–977.
- ^ Maertzdorf J, Repsilber D, Parida SK, Stanley K, Roberts T, Black G, Walzl G, Kaufmann SH. 2011. Human gene expression profiles of susceptibility and resistance in tuberculosis. Genes Immun 12:15–22.
- ^ de Léséleuc L, Orlova M, Cobat A, Girard M, Huong NT, Ba NN, Thuc NV, Truman R, Spencer JS, Adams L, Thai VH, Alcais A, Schurr E. 2013. PARK2 mediates interleukin 6 and monocyte chemoattractant protein 1 production by human macrophages. PLoS Negl Trop Dis 7:e2015.
- ^ Silva BJ, Barbosa MG, Andrade PR, Ferreira H, Nery JA, Côrte-Real S, da Silva GM, Rosa PS, Fabri M, Sarno EN, Pinheiro RO. 2017. Autophagy is an innate mechanism associated with leprosy polarization. PLoS Pathog 13:e1006103.
- ^ Spandl J, Lohmann D, Kuerschner L, Moessinger C, Thiele C. 2011. Ancient ubiquitous protein 1 (AUP1) localizes to lipid droplets and binds the E2 ubiquitin conjugase G2 (Ube2g2) via its G2 binding region. J Biol Chem 286:5599–5606.
- ^ Mattos KA, Oliveira VC, Berrêdo-Pinho M, Amaral JJ, Antunes LC, Melo RC, Acosta CC, Moura DF, Olmo R, Han J, Rosa PS, Almeida PE, Finlay BB, Borchers CH, Sarno EN, Bozza PT, Atella GC, Pessolani MC. 2014. Mycobacterium leprae intracellular survival relies on cholesterol accumulation in infected macrophages: a potential target for new drugs for leprosy treatment. Cell Microbiol 16:797–815.
- ^ Marques MA, Berrêdo-Pinho M, Rosa TL, Pujari V, Lemes RM, Lery LM, Silva CA, Guimarães AC, Atella GC, Wheat WH, Brennan PJ, Crick DC, Belisle JT, Pessolani MC. 2015. The essential role of cholesterol metabolism in the intracellular survival of Mycobacterium leprae is not coupled to central carbon metabolism and energy production. J Bacteriol 197:3698–3707.
- ^ Cruz D, Watson AD, Miller CS, Montoya D, Ochoa MT, Sieling PA, Gutierrez MA, Navab M, Reddy ST, Witztum JL, Fogelman AM, Rea TH, Eisenberg D, Berliner J, Modlin RL. 2008. Host-derived oxidized phospholipids and HDL regulate innate immunity in human leprosy. J Clin Invest 118:2917–2928.
- a, b de Mattos KA, Sarno EN, Pessolani MC, Bozza PT. 2012. Deciphering the contribution of lipid droplets in leprosy: multifunctional organelles with roles in Mycobacterium leprae pathogenesis. Mem Inst Oswaldo Cruz 107(Suppl 1):156–166.
- ^ Tanigawa K, Suzuki K, Nakamura K, Akama T, Kawashima A, Wu H, Hayashi M, Takahashi S, Ikuyama S, Ito T, Ishii N. 2008. Expression of adipose differentiation-related protein (ADRP) and perilipin in macrophages infected with Mycobacterium leprae. FEMS Microbiol Lett 289:72–79.
- ^ Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. 2011. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab 13:655–667.
- ^ Degang Y, Akama T, Hara T, Tanigawa K, Ishido Y, Gidoh M, Makino M, Ishii N, Suzuki K. 2012. Clofazimine modulates the expression of lipid metabolism proteins in Mycobacterium leprae-infected macrophages. PLoS Negl Trop Dis 6:e1936.
- a, b Tanigawa K, Degang Y, Kawashima A, Akama T, Yoshihara A, Ishido Y, Makino M, Ishii N, Suzuki K. 2012. Essential role of hormone-sensitive lipase (HSL) in the maintenance of lipid storage in Mycobacterium leprae-infected macrophages. Microb Pathog 52:285–291.
- a, b Lobato LS, Rosa PS, Ferreira J da S, Neumann Ada S, da Silva MG, do Nascimento DC, Soares CT, Pedrini SC, Oliveira DS, Monteiro CP, Pereira GM, Ribeiro-Alves M, Hacker MA, Moraes MO, Pessolani MC, Duarte RS, Lara FA. 2014. Statins increase rifampin mycobactericidal effect. Antimicrob Agents Chemother 58:5766–5774.
- a, b, c Medeiros RC, Girardi KD, Cardoso FK, Mietto BS, Pinto TG, Gomez LS, Rodrigues LS, Gandini M, Amaral JJ, Antunes SL, Corte-Real S, Rosa PS, Pessolani MC, Nery JA, Sarno EN, Batista-Silva LR, Sola-Penne M, de Oliveira MF, Moraes MO, Lara FA. 2016. Subversion of Schwann cell glucose metabolism by Mycobacterium leprae. J Biol Chem 291:24803.
- ^ Amaral JJ, Antunes LC, de Macedo CS, Mattos KA, Han J, Pan J, Candéa AL, Henriques Md, Ribeiro-Alves M, Borchers CH, Sarno EN, Bozza PT, Finlay BB, Pessolani MC. 2013. Metabonomics reveals drastic changes in anti-inflammatory/pro-resolving polyunsaturated Fatty acids-derived lipid mediators in leprosy disease. PLoS Negl Trop Dis 7:e2381.
- a, b, c Batista-Silva LR, Rodrigues LS, Vivarini Ade C, Costa F da M, Mattos KA, Costa MR, Rosa PS, Toledo-Pinto TG, Dias AA, Moura DF, Sarno EN, Lopes UG, Pessolani MC. 2016. Mycobacterium leprae-induced Insulin-like Growth Factor I attenuates antimicrobial mechanisms, promoting bacterial survival in macrophages. Sci Rep 6:27632.
- a, b Rodrigues LS, da Silva Maeda E, Moreira ME, Tempone AJ, Lobato LS, Ribeiro-Resende VT, Alves L, Rossle S, Lopes UG, Pessolani MC. 2010. Mycobacterium leprae induces insulin-like growth factor and promotes survival of Schwann cells upon serum withdrawal. Cell Microbiol 12:42–54.
- ^ Rodrigues LS, Hacker MA, Illarramendi X, Pinheiro MF, Nery JA, Sarno EN, Pessaloni MC. 2011. Circulating levels of insulin-like growth factor-I (IGF-I) correlate with disease status in leprosy. BMC Infect Dis 11:339.
- ^ Mattos KA, Oliveira VG, D’Avila H, Rodrigues LS, Pinheiro RO, Sarno EN, Pessolani MC, Bozza PT. 2011. TLR6-driven lipid droplets in Mycobacterium leprae-infected Schwann cells: immunoinflammatory platforms associated with bacterial persistence. J Immunol 187:2548–2558.
- ^ Teles RM, Graeber TG, Krutzik SR, Montoya D, Schenk M, Lee DJ, Komisopoulou E, Kelly-Scumpia K, Chun R, Iyer SS, Sarno EN, Rea TH, Hewison M, Adams JS, Popper SJ, Relman DA, Stenger S, Bloom BR, Cheng G, Modlin RL. 2013. Type I interferon suppresses type II interferon-triggered human anti-mycobacterial responses. Science 339:1448–1453.
- a, b Rambukkana A, Yamada H, Zanazzi G, Mathus T, Salzer JL, Yurchenco PD, Campbell KP, Fischetti VA. 1998. Role of alpha-dystroglycan as a Schwann cell receptor for Mycobacterium leprae. Science 282:2076–2079.
- a, b Shimoji Y, Ng V, Matsumura K, Fischetti VA, Rambukkana A. 1999. A 21-kDa surface protein of Mycobacterium leprae binds peripheral nerve laminin-2 and mediates Schwann cell invasion. Proc Natl Acad Sci U S A 96:9857–9862.
- a, b de Lima CS, Marques MA, Debrie AS, Almeira EC, Silva CA, Brennan PJ, Sarno EN, Menozzi FD, Pessolani MC. 2009. Heparin-binding hemagglutinin (HBHA) of Mycobacterium leprae is expressed during infection and enhances bacterial adherence to epithelial cells. FEMS Microbiol Lett 292:162–169.
- a, b Ribeiro-Resende VT, Ribeiro-Guimarães ML, Lemes RM, Nascimento IC, Alves L, Mendez-Otero R, Pessolani MC, Lara FA. 2010. Involvement of 9-O-Acetyl GD3 ganglioside in Mycobacterium leprae infection of Schwann cells. J Biol Chem 285:34086–34096.
- ^ Tapinos N, Ohnishi M, Rambukkana A. 2006. ErbB2 receptor tyrosine kinase signaling mediates early demyelination induced by leprosy bacilli. Nat Med 12:961–966.
- ^ Masaki T, Qu J, Cholewa-Waclaw J, Burr K, Raaum R, Rambukkana A. 2013. Reprogramming adult Schwann cells to stem cell-like cells by leprosy bacilli promotes dissemination of infection. Cell 152:51–67.
- a, b Banerjee S, Farhana A, Ehtesham NZ, Hasnain SE. 2011. Iron acquisition, assimilation and regulation in mycobacteria. Infect Genet Evol 11:825–838.
- ^ Andrews SC. 1998. Iron storage in bacteria. Adv Microb Physiol 40:281–351.
- ^ Hentze MW, Muckenthaler MU, Andrews NC. 2004. Balancing acts: molecular control of mammalian iron metabolism. Cell 117:285–297.
- ^ Macham LP, Ratledge C, Nocton JC. 1975. Extracellular iron acquisition by mycobacteria: role of the exochelins and evidence against the participation of mycobactin. Infect Immun 12:1242–1251.
- ^ Snow GA. 1965. Isolation and structure of mycobactin T, a growth factor from Mycobacterium tuberculosis. Biochem J 97:166–175.
- ^ Gobin J, Moore CH, Reeve JR Jr, Wong DK, Gibson BW, Horwitz MA. 1995. Iron acquisition by Mycobacterium tuberculosis: isolation and characterization of a family of iron-binding exochelins. Proc Natl Acad Sci U S A 92:5189–5193.
- ^ Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT. 1998. Identification of a Mycobacterium tuberculosis gene cluster encoding the biosynthetic enzymes for assembly of the virulence-conferring siderophore mycobactin. Chem Biol 5:631–645.
- ^ De Voss JJ, Rutter K, Schroeder BG, Barry CE. 1999. Iron acquisition and metabolism by mycobacteria. J Bacteriol 181:4443–4451.
- ^ Rodriguez GM, Smith I. 2003. Mechanisms of iron regulation in mycobacteria: role in physiology and virulence. Mol Microbiol 47:1485–1494.
- ^ Li W, He J, Xie L, Chen T, Xie J. 2013. Comparative genomic insights into the biosynthesis and regulation of mycobacterial siderophores. Cell Physiol Biochem 31:1–13.
- ^ Williams DL, Torrero M, Wheeler PR, Truman RW, Yoder M, Morrison N, Bishai WR, Gillis TP. 2004. Biological implications of Mycobacterium leprae gene expression during infection. J Mol Microbiol Biotechnol 8:58–72.
- ^ Cole ST, Eiglmeier K, Parkhill J, James KD, Thomson NR, Wheeler PR, Honoré N, Garnier T, Churcher C, Harris D, Mungall K, Basham D, Brown D, Chillingworth T, Connor R, Davies RM, Devlin K, Duthoy S, Feltwell T, Fraser A, Hamlin N, Holroyd S, Hornsby T, Jagels K, Lacroix C, Maclean J, Moule S, Murphy L, Oliver K, Quail MA, Rajandream MA, Rutherford KM, Rutter S, Seeger K, Simon S, Simmonds M, Skelton J, Squares R, Squares S, Stevens K, Taylor K, Whitehead S, Woodward JR, Barrell BG. 2001. Massive gene decay in the leprosy bacillus. Nature 409:1007–1011.
- ^ Pessolani MC, Smith DR, Rivoire B, McCormick J, Hefta SA, Cole ST, Brennan PJ. 1994. Purification, characterization, gene sequence, and significance of a bacterioferritin from Mycobacterium leprae. J Exp Med 180:319–327.
- ^ Takatsuka M, Osada-Oka M, Satoh EF, Kitadokoro K, Nishiuchi Y, Niki M, Inoue M, Iwai K, Arakawa T, Shimoji Y, Ogura H, Kobayashi K, Rambukkana A, Matsumoto S. 2011. A histone-like protein of mycobacteria possesses ferritin superfamily protein-like activity and protects against DNA damage by Fenton reaction. PLoS One 6:e20985.
- ^ Gruenheid S, Canonne-Hergaux F, Gauthier S, Hackam DJ, Grinstein S, Gros P. 1999. The iron transport protein NRAMP2 is an integral membrane glycoprotein that colocalizes with transferrin in recycling endosomes. J Exp Med 189:831–841.
- ^ Picard V, Govoni G, Jabado N, Gros P. 2000. Nramp 2 (DCT1/DMT1) expressed at the plasma membrane transports iron and other divalent cations into a calcein-accessible cytoplasmic pool. J Biol Chem 275:35738–35745.
- ^ Govoni G, Gros P. 1998. Macrophage NRAMP1 and its role in resistance to microbial infections. Inflamm Res 47:277–284.
- ^ Holmes MA, Paulsene W, Jide X, Ratledge C, Strong RK. 2005. Siderocalin (Lcn 2) also binds carboxymycobactins, potentially defending against mycobacterial infections through iron sequestration. Structure 13:29–41.
- ^ Park CH, Valore EV, Waring AJ, Ganz T. 2001. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem 276:7806–7810.
- ^ Hentze MW, Muckenthaler MU, Galy B, Camaschella C. 2010. Two to tango: regulation of Mammalian iron metabolism. Cell 142:24–38.
- ^ Johnson EE, Wessling-Resnick M. 2012. Iron metabolism and the innate immune response to infection. Microbes Infect 14:207–216.
- ^ Drakesmith H, Prentice AM. 2012. Hepcidin and the iron-infection axis. Science 338:768–772.
- ^ Delaby C, Pilard N, Gonçalves AS, Beaumont C, Canonne-Hergaux F. 2005. Presence of the iron exporter ferroportin at the plasma membrane of macrophages is enhanced by iron loading and down-regulated by hepcidin. Blood 106:3979–3984.
- ^ Theurl I, Theurl M, Seifert M, Mair S, Nairz M, Rumpold H, Zoller H, Bellman-Weiler R, Niederegger H, Talasz H, Weiss G. 2008. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood 111:2392–2399.
- ^ Peyssonnaux C, Zinkernagel AS, Datta V, Lauth X, Johnson RS, Nizet V. 2006. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood 107:3727–3732.
- ^ Oliveira ALG, Chaves AT, Menezes CAS, Guimarães NS, Bueno LL, Fujiwara RT, Rocha MODC. 2017. Vitamin D receptor expression and hepcidin levels in the protection or severity of leprosy: a systematic review. Microbes Infect 19:311–322.
- a, b Souza VN, Malaspina TS, Campanelli AP, Ghidella C, Ura S, Dalpino D, do Nascimento DC, Latini AC. 2012. Increased hepcidin expression in multibacillary leprosy. Mem Inst Oswaldo Cruz 107(Suppl 1):183–189.
- ^ Muruganand D, Daniel E, Ebenezer GJ, Rabboni SE, Segar P, Job CK. 2004. Mycobacterium leprae infection and serum lactoferrin levels. Lepr Rev 75:282–288.
- ^ Momotani E, Wuscher N, Ravisse P, Rastogi N. 1992. Immunohistochemical identification of ferritin, lactoferrin and transferrin in leprosy lesions of human skin biopsies. J Comp Pathol 106:213–220.
- ^ Johnson EE, Srikanth CV, Sandgren A, Harrington L, Trebicka E, Wang L, Borregaard N, Murray M, Cherayil BJ. 2010. Siderocalin inhibits the intracellular replication of Mycobacterium tuberculosis in macrophages. FEMS Immunol Med Microbiol 58:138–145.
- ^ Holmes MA, Paulsene W, Jide X, Ratledge C, Strong RK. 2005. Siderocalin (Lcn 2) also binds carboxymycobactins, potentially defending against mycobacterial infections through iron sequestration. Structure 1993 13:29–41.
- ^ de Mattos Barbosa MG, da Silva Prata RB, Andrade PR, Ferreira H, de Andrade Silva BJ, da Paixão de Oliveira JA, Assis TQ, de Toledo-Pinto TG, de Lima Bezerra OC, da Costa Nery JA, Rosa PS, Bozza MT, Lara FA, Moraes MO, Schmitz V, Sarno EN, Pinheiro RO. 2017. Indoleamine 2,3-dioxygenase and iron are required for Mycobacterium leprae survival. Microbes Infect doi: 10.1016/j.micinf.2017.06.006
- ^ Souza VN, Malaspina TS, Campanelli AP, Ghidella C, Ura S, Dalpino D, do Nascimento DC, Latini AC. 2012. Increased hepcidin expression in multibacillary leprosy. Mem Inst Oswaldo Cruz 107(Suppl 1):183–189.
- ^ Sher R, Shulman G, Baily P, Politzer WM. 1981. Serum trace elements and vitamin A in leprosy subtypes. Am J Clin Nutr 34:1918–1924.
- ^ Lapinsky SE, Baynes RD, Schulz EJ, MacPhail AP, Mendelow B, Lewis D, Bothwell TH. 1992. Anaemia, iron-related measurements and erythropoietin levels in untreated patients with active leprosy. J Intern Med 232:273–278.
- ^ Jain A, Mukherjee A, Chattopadhya D, Saha K. 1995. Biometals in skin and sera of leprosy patients and their correlation to trace element contents of M. leprae and histological types of the disease; a comparative study with cutaneous tuberculosis. Int J Lepr Other Mycobact Dis 63:249–258.
- ^ Rea TH. 2001. Decreases in mean hemoglobin and serum albumin values in erythema nodosum leprosum and lepromatous leprosy. Int J Lepr Other Mycobact Dis 69:318–327.
- a, b, c, d, e, f Moura DF, de Mattos KA, Amadeu TP, Andrade PR, Sales JS, Schmitz V, Nery JA, Pinheiro RO, Sarno EN. 2012. CD163 favors Mycobacterium leprae survival and persistence by promoting anti-inflammatory pathways in lepromatous macrophages. Eur J Immunol 42:2925–2936.
- ^ Tullius MV, Harmston CA, Owens CP, Chim N, Morse RP, McMath LM, Iniguez A, Kimmey JM, Sawaya MR, Whitelegge JP, Horwitz MA, Goulding CW. 2011. Discovery and characterization of a unique mycobacterial heme acquisition system. Proc Natl Acad Sci U S A 108:5051–5056.
- ^ Jones CM, Niederweis M. 2011. Mycobacterium tuberculosis can utilize heme as an iron source. J Bacteriol 193:1767–1770.
- ^ Elamin AA, Stehr M, Singh M. 2012. Lipid Droplets and Mycobacterium leprae Infection. J Pathog 2012:361374. doi: 10.1155/2012/361374
- ^ Murphy DJ. 2001. The biogenesis and functions of lipid bodies in animals, plants and microorganisms. Prog Lipid Res 40:325–438.
- ^ Melo RCN, Dvorak AM. 2012. Lipid body-phagosome interaction in macrophages during infectious diseases: host defense or pathogen survival strategy? PLoS Pathog 8:e1002729.
- a, b Luo M, Fadeev EA, Groves JT. 2005. Mycobactin-mediated iron acquisition within macrophages. Nat Chem Biol 1:149–153.
- ^ Wang TT, Nestel FP, Bourdeau V, Nagai Y, Wang Q, Liao J, Tavera-Mendoza L, Lin R, Hanrahan JW, Mader S, White JH. 2004. Cutting edge: 1,25-dihydroxyvitamin D3 is a direct inducer of antimicrobial peptide gene expression. J Immunol 173:2909–2912.
- ^ Martineau AR, Wilkinson KA, Newton SM, Floto RA, Norman AW, Skolimowska K, Davidson RN, Sørensen OE, Kampmann B, Griffiths CJ, Wilkinson RJ. 2007. IFN-gamma- and TNF-independent vitamin D-inducible human suppression of mycobacteria: the role of cathelicidin LL-37. J Immunol 178:7190–7198.
- ^ Liu PT, Stenger S, Tang DH, Modlin RL. 2007. Cutting edge: vitamin D-mediated human antimicrobial activity against Mycobacterium tuberculosis is dependent on the induction of cathelicidin. J Immunol 179:2060–2063.
- ^ Liu PT, Schenk M, Walker VP, Dempsey PW, Kanchanapoomi M, Wheelwright M, Vazirnia A, Zhang X, Steinmeyer A, Zügel U, Hollis BW, Cheng G, Modlin RN. 2009. Convergence of IL-1beta and VDR activation pathways in human TLR2/1-induced antimicrobial responses. PLoS One 4:e5810.
- ^ de Sousa JR, de Sousa RP, Aarão TL, Dias LB Jr, Carneiro FR, Fuzii HT, Quaresma JA. 2016. In situ expression of M2 macrophage subpopulation in leprosy skin lesions. Acta Trop 157:108–114.
- ^ Chung AW, Sieling PA, Schenk M, Teles RM, Krutzik SR, Hsu DK, Liu FT, Sarno EN, Rea TH, Stenger S, Modlin RL, Lee DJ. 2013. Galectin-3 regulates the innate immune response of human monocytes. J Infect Dis 207:947–956.
- a, b Yang D, Shui T, Miranda JW, Gilson DJ, Song Z, Chen J, Shi C, Zhu J, Yang J, Jing Z. 2016. Mycobacterium leprae-infected macrophages preferentially primed regulatory T cell responses and was associated with lepromatous leprosy. PLoS Negl Trop Dis 10:e0004335.
- ^ Pereira-Suárez AL, Alvarado-Navarro A, Barrietos-García JG, Estrada-Chávez C, Muñoz-Valle JF, Fafutis-Morris M. 2015. Differential expression of solute carrier family 11a member 1 and inducible nitric oxide synthase 2 in skin biopsies from leprosy patients. Indian J Dermatol Venereol Leprol 81:594–599.
- ^ de Souza Sales J, Lara FA, Amadeu TP, de Oliveira Fulco T, da Costa Nery JA, Sampaio EP, Pinheiro RO, Sarno EN. 2011. The role of indoleamine 2, 3-dioxygenase in lepromatous leprosy immunosuppression. Clin Exp Immunol 165:251–263.
- ^ Weaver LK, Hintz-Goldstein KA, Pioli PA, Wardwell K, Qureshi N, Vogel SN, Guyre PM. 2006. Pivotal advance: activation of cell surface toll-like receptors causes shedding of the hemoglobin scavenger receptor CD163. J Leukoc Biol 80:26–35.
- ^ Schaer CA, Schoedon G, Imhof A, Kurrer MO, Schaer DJ. 2006. Constitutive endocytosis of CD163 mediates hemoglobin-heme uptake and determines the noninflammatory and protective transcriptional response of macrophages to hemoglobin. Circ Res 99:943–950.
- ^ Alcaraz MJ, Fernández P, Guillén MI. 2003. Anti-inflammatory actions of the heme oxygenase-1 pathway. Curr Pharm Des 9:2541–2551.
- a, b Sinsimer D, Fallows D, Peixoto B, Krahenbuhl J, Kaplan G, Manca C. 2010. Mycobacterium leprae actively modulates the cytokine response in naïve human monocytes. Infect Immun 78:293–300.
- ^ Teles RMB, Kelly-Scumpia KM, Sarno EN, Rea TH, Ochoa MT, Cheng G, Modlin RL. 2015. IL-27 suppresses antimicrobial activity in human leprosy. J Invest Dermatol 135:2410–2417.
- ^ Weiss G, Schaible UE. 2015. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev 264:182–203.
- ^ de Oliveira Fulco T, Andrade PR, de Mattos Barbosa MG, Pinto TG, Ferreira PF, Ferreira H, da Costa Nery JA, Real SC, Borges VM, Moraes MO, Sarno EN, Sampaio EP, Pinheiro RO. 2014. Effect of apoptotic cell recognition on macrophage polarization and mycobacterial persistence. Infect Immun 82:3968–3978.